Epidemiología de las miocardiopatías hereditarias
- Dra Josefina Parodi
- diciembre 23, 2020
- Consejo Cardiomiopatías y Enfermedades del Pericardio
- 0 Comments
La siguiente revisión de McKenna y Judge intenta explicar las particularidades diagnósticas de las distintas miocardiopatías, abordándolas desde su epidemiología, clínica y genética.
A diferencia de otras enfermedades cardiovasculares, los distintos tipos de miocardiopatías, sea hipertrófica, dilatada, arrítmica o restrictiva, no tienen una clara descripción de su incidencia y prevalencia reales, e incluso en la mayoría de los artículos y guías de manejo clínico no se discute esta temática en profundidad. Los escasos datos disponibles provienen principalmente de estudios de pesquisa poblacionales, incluso en muchos casos obtenidos mediante extrapolación de datos (1).
Actualmente se considera que la prevalencia en adultos de Miocardiopatía Hipertrófica (MCH) es de 1 en 500 individuos, de Miocardiopatía Dilatada (MCD) es de 1 en 250, de Miocardiopatía Arritmogénica (MCA) es de 1 en 5000, y la de miocardiopatía restrictiva es rara. Las miocardiopatías hereditarias han tomado relevancia clínica en los últimos años, principalmente por su potencialidad de generar insuficiencia cardíaca y muerte súbita a edades tempranas. Esto ha motivado el surgimiento de nuevas herramientas diagnósticas e investigaciones en el área, logrando un mejor conocimiento de las mutaciones genéticas potencialmente patogénicas que caracterizan a la enfermedad (2). No obstante, el completo entendimiento de las miocardiopatías y la caracterización de sus variantes genéticas sigue siendo un desafío.
La siguiente revisión de McKenna y Judge intenta explicar las particularidades diagnósticas de las distintas miocardiopatías, abordándolas desde su epidemiología, clínica y genética, con el objetivo de clarificar el conocimiento sobre su herencia, presentación clínica y evolución.
MIOCARDIOPATÍA HIPERTRÓFICA
El diagnóstico de MCH se basa en la presencia de ≥15 mm de diámetro parietal en al menos un segmento ventricular izquierdo (VI) por Ecodoppler transtorácico (ETT) o Resonancia Magnética Nuclear (RMN), no explicado por otra causa como hipertensión arterial (HTA), enfermedad coronaria o valvular (Fig.1), corregido por superficie corporal (SC) en niños (3). Hasta el 95% de los pacientes con MCH tienen alteraciones en el electrocardiograma (ECG), típicamente eje desviado a izquierda, ondas Q patológicas, alteraciones en segmento ST, u ondas T negativas (4).
El primer dilema lo encontramos en la definición de MCH, siendo únicamente para algunos autores aquellas cuya fisiopatología está asociada a la presencia de variantes en genes que codifican para proteínas sarcoméricas, mientras que otros autores incluyen en esta clasificación enfermedades no sarcoméricas como la amiloidosis o enfermedad de Fabry (5).
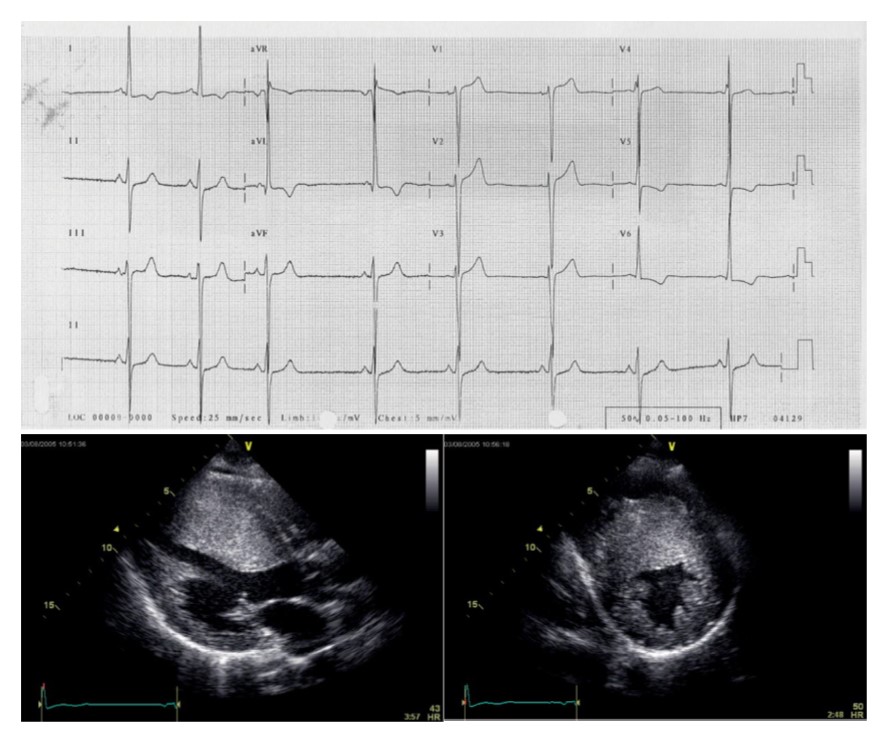
Fig. 1. Paciente masculino de 19 años con MCH. A) ECG 12 derivaciones con cambios difusos de la repolarización. B) Ecocardiograma 2D en eje largo (derecha) y corto (izquierda) paraesternal mostrando hipertrofia de ventrículo izquierdo de 30 mm de máximo espesor.
Respecto a su prevalencia, la mayor parte de los datos disponibles provienen de estudios de screening en poblaciones generales, en atletas de alto rendimiento, o en reclutas militares. Invariablemente en casi todos estos estudios la presencia de HTA aunque leve era considerada suficiente para explicar los hallazgos electrocardiográficos y por ende excluir los pacientes de la probable prevalencia de MCH. No obstante, sabemos que la HTA salvo caso severa, de larga data y no tratada, raramente produce cambios en ECG o hipertrofia ventricular >2 cm en ETT. Por lo tanto, no incluir estos pacientes significa automáticamente una subestimación de la prevalencia de MCH, considerándose hasta del 30%. Respecto a sus diferencias epidemiológicas, el estudio CARDIA (6) mostró una prevalencia del 0.17%, siendo esta levemente mayor en hombres (0.26%) y raza negra (0.24%) respecto a mujeres (0.09%) y caucásicos (0.10%). De los categorizados como portadores de MCH, aproximadamente el 60% refiere antecedentes familiares de cardiopatía, presentando estos pacientes mejor pronóstico y menos probabilidad de tener variantes patogénicas de proteínas sarcoméricas (7).
La prevalencia de MCH en pacientes pediátricos se encuentra mejor registrada en importantes bases de datos nacionales, como en las de Estados Unidos, Australia y Canadá, reportando una incidencia general de 0.47/100000 niños, levemente más frecuente en hombres y en niños <1 año. En un tercio de los casos se describen secundarios a causas diferentes a la mutación de proteínas sarcoméricas: errores del metabolismo, malformaciones cardiacas, o enfermedades neuromusculares (8,9).
La epidemiología genética de la MCH se encuentra mejor descripta que la de otras cardiopatías, presentando el 60% de los pacientes con formas hereditarias mutaciones en los genes que codifican proteínas sarcoméricas. Más del 80% de estas corresponden a las variantes MYBPC3 y MYH7, que codifican la proteína C fijadora de miosina y la de cadena pesada de miosina respectivamente (10). A diferencia de la población general, la prevalencia en familiares de pacientes con MCH diagnosticada es de hasta 1 cada 2, considerando en este número también la posibilidad de expresiones incompletas de la enfermedad, es decir, típicamente manifestando un ECG patológico pero sin hipertrofia en ETT. Esto es más frecuentemente observado en pacientes portadores de variantes en el gen de la troponina I (TNNI3). Asimismo existen pacientes con formas tardías de presentación, que suelen ser portadores de la variante MYBPC3. En todos los casos, a la hora de observar un ECG patológico compatible con MCH en ausencia de hipertrofia en ETT, es importante considerar la posibilidad de tratarse de una MCH con fenotipo incompleto, en lugar de considerarse secundario a HTA o al ejercicio extremo. En caso de duda, la reversión de la hipertrofia mediante tratamiento de la HTA o descenso del monto de actividad física puede clarificar la situación.
Mención especial requieren los pacientes con hipertrofia septal basal aislada, donde la posibilidad de ser portadores de una mutación genética es mucho más baja, así como menor incidencia de obstrucción del tracto de salida de VI o de fibrosis en la RMN (11). Incluso muchos autores actualmente consideran a la hipertrofia septal basal (clásicamente conocida como septum sigmoideo) como una consecuencia normal de la edad.
MIOCARDIOPATÍA DILATADA
Aún más complicada es la descripción epidemiológica de la miocardiopatía dilatada, ya que algunos autores incluyen en su definición solamente a las secundarias a causas hereditarias, clásicamente interpretadas como “idiopáticas”, mientras que otros las dividen en de origen isquémico o no isquémico (3, 12).
Se define como MCD a la presencia de disfunción con dilatación ventricular izquierda no explicada por enfermedad coronaria, valvular o hipertensiva, entendiendo como disfunción ventricular la presencia de una fracción de eyección <50% por ETT, tomografía cardíaca (CT) o RMN (13). Pero la definición de dilatación ventricular resulta más compleja, ya que debe ajustarse a la superficie corporal: para los autores, se considera a un diámetro de fin de diástole (DFD) >2 desvíos estándar de la población normal por TC o RMN, y >112% del DFD esperado según la Fórmula de Henry: [DDVI = (45.3 × BSA0.3) – (0.03 × edad) − 7.2] (14).
No sólo su definición es materia de debate; no existe ningún estudio epidemiológico que haya analizado su real incidencia y prevalencia, sino que la misma proviene de estimaciones de estudios poblacionales, como el estudio Olmested County (15) donde se observó una incidencia de 1/2700 pacientes. En este estudio, la incidencia de MCH fue de 1/5100 individuos; al reconocerse en estudios posteriores que esta incidencia se encuentra infraestimada aproximadamente 10 veces, es así que se llegó a la conclusión que la incidencia de MCD es de aproximadamente 1/250 en lugar de 1/2700. Es decir, una aseveración aunque teóricamente correcta, poco fundada. Estudios posteriores encontraron similares valores que podrían avalar esta teoría; por ejemplo el estudio MESA (16) reveló una prevalencia de disfunción ventricular asintomática idiopática por RMN en 1.7%.
La información epidemiológica en pacientes pediátricos es un poco más robusta. En los mismos registros de Australia y USA de MCH (8-9), se observó una incidencia anual de MCD en 0.73/100000 y 0.53/100000 niños <18 años respectivamente, en su mayoría de origen idiopático (66%), en segundo lugar por miocarditis (16%), y patologías neuromusculares (9%). Se observó historia familiar de MCD en 15-20% de estos pacientes.
Respecto a la penetrancia del genotipo patológico, registros de pacientes trasplantados por MCD en USA han revelado presencia de familiares afectados hasta en el 26% de los casos (17). Se considera que cuando se encuentra un paciente con MCD se debe estudiar a tres generaciones anteriores, encontrando según los datos disponibles al menos un miembro de la familia con disfunción ventricular en hasta 37% de los casos, y/o con dilatación ventricular en hasta 50% (18). Otros tipos de expresiones incompletas de la enfermedad incluyen trastornos aislados de conducción, reducción de la capacidad de ejercicio, entre otros fenotipos.
La gran heterogeneidad fenotípica y epidemiológica de la MCD va de la mano con su dificultad de diagnóstico genético. A diferencia de las otras miocardiopatías, existen descriptos >60 genes relacionados con la MCD (19), siendo los más frecuentes involucrados los que codifican proteínas del sarcómero o laminina A/C. Variantes en el gen que codifica titina se identifica en 20-30% de los pacientes, pero la misma puede encontrarse en hasta un 3% de pacientes sanos por lo que su significancia patológica todavía se encuentra en discusión.
MIOCARDIOPATIA ARRITMOGÉNICA
Similar a lo que ocurre con la MCD, algunos autores creen que cualquier tipo de miocardiopatía con propensión a producir arritmia, por ejemplo sarcoidosis o enfermedades infiltrativas, deberían incluirse en esta categoría. No obstante, el grupo de McKenna utiliza este epónimo para describir a la enfermedad antiguamente conocida como displasia arritmogénica del ventrículo derecho (DAVD), enfermedad caracterizada por afección principalmente del ventrículo derecho (VD) salvo etapas avanzadas, con dilatación y disfunción del VD y reemplazo de miocitos de la pared libre por tejido cicatrizal predominantemente de tipo fibroadiposo (20). Hoy en día se sabe que esta displasia puede afectar tanto al ventrículo derecho como al izquierdo, y es por eso que se denomina actualmente Miocardiopatía arritmogénica; si bien este término puede incluir otras miocardiopatías hereditarias con tendencia a disfunción ventricular y arritmias, es claro que la DAVD es su máximo exponente (21).
La DAVD se define por la presencia de criterios mayores y menores dentro de los antecedentes familiares, los hallazgos en ECG, las manifestaciones arrítmicas, las alteraciones del VD (ajustadas por superficie corporal) en estudios de imágenes, y los hallazgos histopatológicos (22). Su prevalencia es incierta. Estudios iniciales en territorios italianos como en la región de Veneto parecían reclamar que esta arritmia era más prevalente en países Europeos, principalmente en Italia (23). No obstante, posterior evidencia demostró que si bien la mayoría de los casos están relacionados con una mutación genética fundadora similar, existen distintos tipos de variantes en distintas regiones del mundo capaces de manifestarse como MCA. La prevalencia calculada es de aproximadamente 1/2000 o 1/5000, pero un grupo Alemán reportó una incidencia en su población de hasta 1/1000 (24).
Los genotipos asociados a la MCA se caracterizan por poseer una penetrancia relativamente baja, y fenotípicamente cerca de un tercio de los pacientes presenta expresión completa, un tercio expresión incompleta, y un tercio son portadores sanos. La edad de presentación suele ser entre la segunda y tercera década de la vida, siendo muy rara antes de los 10 años (25). Se sabe que la actividad física juega un rol importante en la patogenia de la enfermedad (26), observando manifestaciones más precoces y más graves en pacientes que realizan más actividad aeróbica, ya que el ejercicio genera desproporcionadamente mayor estiramiento en el VD que en el VI, favoreciendo al daño en los desmosomas y por ende una peor evolución de la enfermedad. Se cree que otros factores ambientales aún desconocidos deberían jugar un rol en la manifestación clínica de la MCA, dado que la mayoría de las variantes encontradas son mutaciones fundadoras, y la incidencia de variantes patogénicas de novo es muy baja.
Aproximadamente el 60% de las MCA son causadas por mutaciones en el gen que codifica proteínas del desmosoma, siendo la variante PKP2 que codifica la proteína placofilina 2 la más reportada (21). Otras variantes frecuentemente reportadas se encuentran en los genes que codifican la desmogleína 2 (DSG2), y desmocolina 2 (DSC2). Las conocidas enfermedades de Naxos y Caravajal son causadas por variantes autosómicas recesivas de los genes de placoglobina y desmoplaquina respectivamente (27). Estas se caracterizan por producir típicamente un cuadro de MCA asociado a alteraciones en piel (queratodermia palmoplantar y cabello lanudo), dado que los desmosomas se encuentran también en estos tejidos. Junto con las tres variantes autosómicas dominantes previamente nombradas, estos 5 genes son responsables de la mayoría de los casos de MCA.
Existen además mutaciones no desmosomales descriptas en pacientes con MCA que presentan entrecruzamiento con otros tipos de miocardiopatías, como la MCD. Mutaciones de SCN5A correspondientes al canal de sodio, asociada a Síndrome de QT largo tipo 3 en caso de pérdida de función o a Síndrome de Brugada en caso de ganancia de función, se han visto asociadas a MCA tanto ventricular izquierda como derecha (28).
MIOCARDIOPATÍA RESTRICTIVA
La miocardiopatía restrictiva es definida por la presencia de patrón de llenado ventricular restrictivo en ETT o hemodinamia, con función y diámetros ventriculares normales, y ausencia de hipertrofia de VI (3). Si bien se han descripto variantes patogénicas propias de esta enfermedad, en la mayor parte de los casos se tratan de fenotipos especiales de otras miocardiopatías, principalmente la MCH (29), salvo la asociada a amiloidosis por mutación del gen de transtirretina. La presencia de MCR hereditaria aislada en pacientes sin antecedentes familiares de otro fenotipo de miocardiopatía es extremadamente raro.
La revisión culmina con un comentario acerca del miocardio no compacto, el cual se define actualmente por la presencia de un radio de tejido no compacto/compacto >2.3 del ventrículo izquierdo en fin de diástole por RMN (30). Un importante metaanálisis plantea que su presencia, hoy quizás sobrediagnosticada gracias a los avances en métodos de diagnóstico por imágenes, no necesariamente conlleva una significancia clínica, y, en ausencia de disfunción ventricular, puede no acarrear mal pronóstico, incluso pudiendo significar una adaptación fisiológica (31).
En resumen, la prevalencia de MCH en adultos es aproximadamente 1/500 con cierta evidencia robusta de ello, y es menos frecuente en poblaciones pediátricas, siendo más común en pacientes <1 año y generalmente asociado a otras enfermedades como alteraciones del metabolismo o patologías neuromusculares, más que consecuencia de mutaciones de proteínas sarcoméricas, como lo es en el 60% de los casos en los adultos. Los fenotipos o manifestaciones clínicas pueden abarcar desde expresiones incompletas (sólo hallazgos patológicos en ECG) hasta debutar como muerte súbita. La evidencia sobre la prevalencia de MCD, considerada 1/250, se genera a raíz de extrapolar resultados, cuya interpretación debe ser tomada con cautela. La incidencia en población pediátrica se encuentra mejor estudiada, siendo ésta el doble de frecuente que la MCH. La expresividad de la MCD es muy variable, siendo más frecuentes las formas incompletas y tardías, incluso hasta la sexta década de vida. La MCA y la miocardiopatía arritmogénica del VD presentan menor prevalencia, aproximadamente 1/5000, pero su potencial debut con muerte súbita como primera manifestación y la relación de su expresión clínica con el ejercicio, obliga al médico a conocerla en profundidad. Las mutaciones en proteínas del desmosoma explican más del 60% de los casos. Finalmente, la MCR familiar es una entidad infrecuente, generalmente causada por variantes genéticas responsables habitualmente de otras miocardiopatías, y el MNC se debe considerar actualmente un rasgo más que una miocardiopatía en sí.
La MCH, la MCD y la MCA son enfermedades con patrón de herencia mendeliana, es decir, causadas por la mutación o alteración en la secuencia de ADN de un solo gen que se hereda de forma directa; en todos los casos predominan las variantes patogénicas autosómicas dominantes, pero con una baja penetrancia. No obstante, existe una discordancia entre la frecuencia de los alelos heredados y la manifestación fenotípica de la enfermedad, principalmente cuando se trata de la MCD o la MCA. Una mejor comprensión de la influencia de los modificadores tanto genéticos como ambientales es necesaria.
La revisión de McKenna analiza en forma exhaustiva todas las prevalencias reportadas y todas las variantes patogénicas (y aquellas de patogenicidad incierta) halladas por diferentes autores a lo largo y ancho del mapa, con el objetivo de resumir la evidencia actual de la epidemiología de este grupo de enfermedades, que claramente sigue siendo un enigma. Deja en evidencia la presencia de un claro desafío para el diagnóstico de las miocardiopatías hereditarias: no solo por la gran variabilidad de fenotipos posibles encontrados (desde cuadros clínicos completos y floridos hasta variantes leves y muchas veces subdiagnosticadas) y el entrecruzamiento de genotipos, sino además por el limitado entendimiento por parte del cardiólogo clínico, en algún punto materia de unos pocos especialistas que acumulan estudios bajo su misma autoría.
La importancia de conocer en más profundidad estas miocardiopatías radica en la potencialidad de presentarse con muerte súbita como debut clínico, y su tendencia a afectar pacientes en décadas más tempranas de la vida, considerados por lo demás sanos desde el punto de vista cardiovascular.
Como mensaje al médico no especialista, es importante siempre recordar que el algoritmo diagnóstico debería incluir además del ECG, el estudio por imágenes e idealmente la evaluación o al menos el asesoramiento genético, la evaluación exhaustiva a familiares, y no solamente de primer grado. La presencia de un ECG compatible con por ejemplo una MCH aún en ausencia de hipertrofia ventricular, no debería considerarse como un hallazgo o una respuesta anormal al ejercicio o a la HTA, sino como un paciente portador de MCH con fenotipo incompleto.
No obstante, la realización rutinaria de estudios genéticos sigue siendo materia de debate y la interpretación de sus hallazgos un desafío, entendiendo que existen variantes de genes también presentes en poblaciones sanas, sin implicancias patológicas.
El gran valor de esta revisión radica en que un mejor entendimiento de las características epidemiológicas y genéticas debería contribuir en última instancia a una mejor comprensión, diagnóstico y tratamiento de las miocardiopatías hereditarias.
Ver link AQUI
BIBLIOGRAFIA


{kind=link}