Más allá de la fracción de eyección: hacia un nuevo paradigma de integración entre la genética y la resonancia en la miocardiopatía dilatada
- Dr. Gabriel Parma
- junio 17, 2026
- Consejo Cardiopatías Congénitas y Pediatría, Editoriales
- Consejo miocardiopatías, insuficiencia cardíaca, miocardiopatía dilatada, miocardiopatias, muerte súbita
- 0 Comments
Introducción
La miocardiopatía dilatada (MCD) continúa siendo una de las principales causas de insuficiencia cardíaca y muerte súbita en adultos jóvenes. Sin embargo, su abordaje clínico ha estado históricamente centrado en la fracción de eyección del ventrículo izquierdo (FEVI), un modelo que ha demostrado claras limitaciones. La evidencia contemporánea muestra que la MCD no es una entidad única, sino un espectro heterogéneo de condiciones con bases genéticas, inflamatorias y ambientales, donde el riesgo arrítmico y la progresión clínica no pueden explicarse únicamente por la disfunción sistólica.
En este contexto, la integración entre genética cardiovascular y resonancia magnética cardíaca (RMC) representa un cambio de paradigma en evolución, orientado hacia una medicina de precisión.
El fracaso del modelo centrado en la FEVI
La dependencia exclusiva de la FEVI como marcador de riesgo ha sido progresivamente cuestionada. Un número significativo de pacientes con FEVI moderadamente reducida o incluso preservada presentan muerte súbita como primera manifestación, mientras que otros con disfunción severa permanecen estables durante años.
Este enfoque ignora el sustrato arrítmico real de la enfermedad, que reside en la arquitectura miocárdica y su determinante biológico. La ausencia de caracterización etiológica y tisular conduce a una estratificación incompleta y a decisiones terapéuticas subóptimas.
Genética: del diagnóstico etiológico a la estratificación de riesgo
La genética cardiovascular ha transformado la comprensión de la MCD. Se reconoce que hasta un 30–40% de los casos tienen una base hereditaria, y que determinadas mutaciones confieren un riesgo arrítmico elevado independiente de la FEVI.
Mutaciones en genes como LMNA, FLNC, RBM20, PLN y DSP identifican fenotipos con alta inestabilidad eléctrica. En estos casos, la indicación de cardiodesfibrilador implantable (CDI) puede considerarse en fases más precoces.
Por otro lado, variantes en TTN, BAG3 o MYH7 se asocian con fenotipos predominantemente de insuficiencia cardíaca, aunque con una heterogeneidad significativa modulada por factores ambientales y comorbilidades.
Resonancia magnética cardíaca: el sustrato como eje
La RMC ha consolidado su rol en la caracterización de la MCD. El realce tardío con gadolinio (LGE) permite identificar fibrosis miocárdica, un marcador robusto e independiente de eventos arrítmicos y mortalidad.
Técnicas como el mapeo T1 y la cuantificación del volumen extracelular permiten detectar fibrosis difusa en estadios tempranos. En este contexto, el edema miocárdico puede reflejar actividad inflamatoria y asociarse a peor pronóstico en determinados escenarios clínicos.
Genotipo + fenotipo: el nuevo paradigma
La integración de la información genética y la caracterización tisular mediante RMC permite una estratificación de riesgo más precisa. Existe evidencia creciente de correlación entre genotipo y patrones de fibrosis.
Desde una perspectiva clínica, se ha propuesto que ciertas mutaciones pudieran asociarse a patrones específicos de fibrosis, constituyendo una hipótesis emergente que vincula genotipo y sustrato estructural.
Este modelo redefine la evaluación de la MCD, donde la FEVI pasa a ser un parámetro más dentro de un enfoque multidimensional.
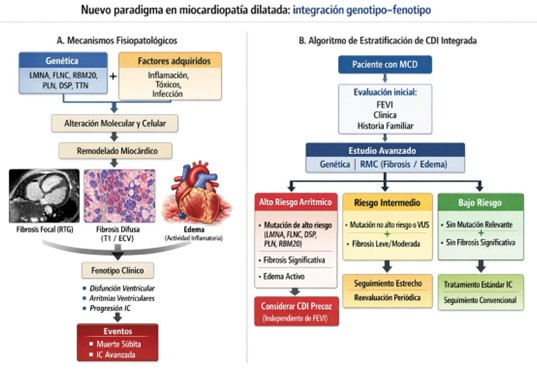
Figura 1. Nuevos paradigmas (modelo integrador) en miocardiopatía dilatada. A: Mecanismos de integración basados en aspectos fisiopatológicos B: Algoritmos de re-estratificación.
Implicancias clínicas: hacia una medicina de precisión
La adopción de este modelo tiene implicancias directas en la práctica clínica:
- Identificación precoz de pacientes de alto riesgo arrítmico
- Indicación más precisa de CDI
- Estratificación basada en sustrato y genética
- Optimización del seguimiento
- Integración del screening familiar
TTN: un modelo particular
TTN merece una consideración particular por su alta prevalencia. Aunque clásicamente se ha asociado a evolución más lenta y menor riesgo arrítmico inicial, la evidencia actual demuestra que no debe considerarse una forma benigna.
Se trata de una entidad heterogénea, frecuentemente asociada a insuficiencia cardíaca y modulada por factores ambientales (“second hits”).
En este contexto, la RMC adquiere un rol central en la estratificación del riesgo, especialmente mediante la detección de fibrosis o inflamación.
El riesgo arrítmico tiende a aumentar con la progresión de la enfermedad, reforzando la necesidad de un enfoque integrador.
Desafíos actuales
A pesar de la evidencia, existen barreras para su implementación:
- Acceso limitado a estudios genéticos y RMC
- Complejidad en la interpretación genética
- Limitada incorporación en guías clínicas
- Necesidad de formación específica
Conclusión
La miocardiopatía dilatada debe entenderse como una enfermedad de sustrato más que exclusivamente funcional. La FEVI, aunque útil, es insuficiente como único marcador.
La integración entre genética y RMC permite una aproximación más precisa e individualizada. El desafío actual no es demostrar este cambio, sino implementarlo en la práctica clínica.
Bibliografía
- Elliott P, et al. 2014 ESC Guidelines on diagnosis and management of cardiomyopathies. Eur Heart J.
- Gulati A, et al. Association of fibrosis with mortality in dilated cardiomyopathy.
- Halliday BP, et al. Prognostic value of LGE in DCM.
- Van Rijsingen IA, et al. Risk stratification in LMNA cardiomyopathy. J Am Coll Cardiol.
- Towbin JA, et al.2025. Genetic basis of dilated cardiomyopathy.
- Angriman, F. Et al 2025. Titin Cardiomyopathy, Emerging Evidence: More Than A Big Heart. Current Cardiology Reports

{kind=link}